Mutect2 joint somatic variant calling workflow (CRAM)¶
Mutect2JointSomaticWorkflowCram · 1 contributor · 1 version
- This workflow uses the capability of mutect2 to call several samples at the same time and improve recall and accuracy through a joint model.
- Most of these tools are still in a beta state and not intended for main production (as of 4.1.4.0) There are also som major tweaks we have to do for runtime, as the amount of data might overwhelm the tools otherwise.
Quickstart¶
from janis_bioinformatics.tools.dawson.workflows.variantcalling.multisample.mutect2.mutect2jointsomaticworkflow_cram import Mutect2JointSomaticWorkflowCram wf = WorkflowBuilder("myworkflow") wf.step( "mutect2jointsomaticworkflowcram_step", Mutect2JointSomaticWorkflowCram( normalBams=None, tumorBams=None, normalName=None, biallelicSites=None, reference=None, panelOfNormals=None, germlineResource=None, ) ) wf.output("out", source=mutect2jointsomaticworkflowcram_step.out)
OR
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
- Generate user input files for Mutect2JointSomaticWorkflowCram:
# user inputs
janis inputs Mutect2JointSomaticWorkflowCram > inputs.yaml
inputs.yaml
biallelicSites: biallelicSites.vcf.gz
germlineResource: germlineResource.vcf.gz
normalBams:
- normalBams_0.cram
- normalBams_1.cram
normalName: <value>
panelOfNormals: panelOfNormals.vcf.gz
reference: reference.fasta
tumorBams:
- tumorBams_0.cram
- tumorBams_1.cram
- Run Mutect2JointSomaticWorkflowCram with:
janis run [...run options] \
--inputs inputs.yaml \
Mutect2JointSomaticWorkflowCram
Information¶
URL: No URL to the documentation was provided
| ID: | Mutect2JointSomaticWorkflowCram |
|---|---|
| URL: | No URL to the documentation was provided |
| Versions: | 0.1.1 |
| Authors: | Sebastian Hollizeck |
| Citations: | |
| Created: | 2019-10-30 |
| Updated: | 2020-12-10 |
Outputs¶
| name | type | documentation |
|---|---|---|
| out | Gzipped<VCF> |
Workflow¶
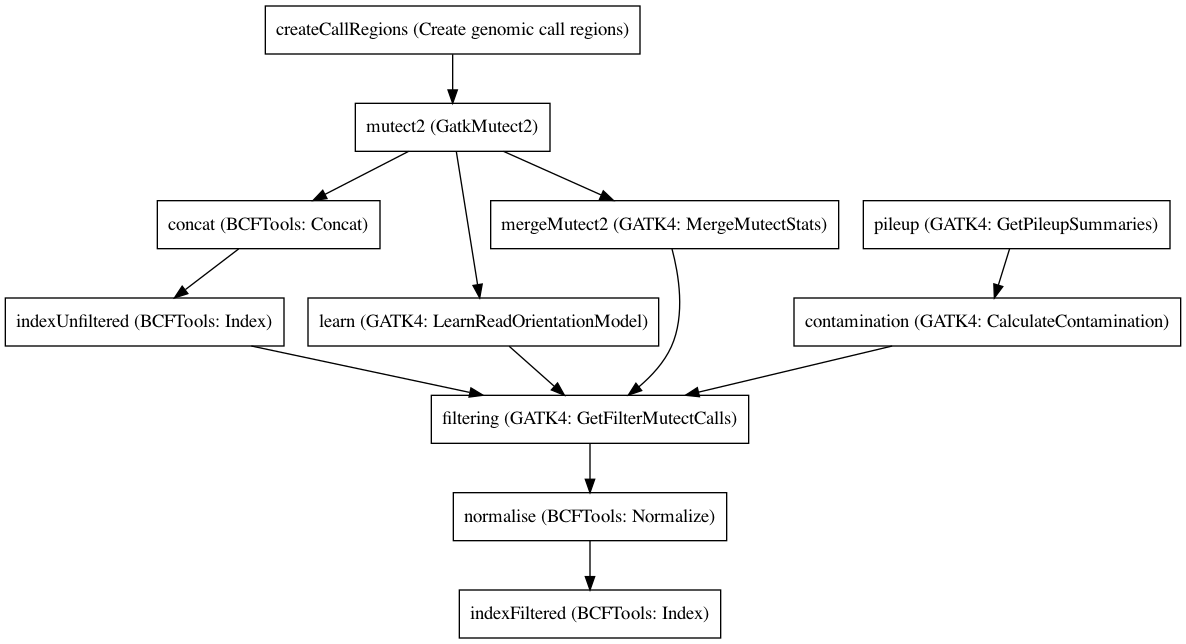
Embedded Tools¶
| Create genomic call regions | CreateCallRegions/v0.1.0 |
| GatkMutect2 | Gatk4Mutect2_cram/4.1.8.1 |
| BCFTools: Concat | bcftoolsConcat/v1.9 |
| BCFTools: Index | bcftoolsIndex/v1.9 |
| GATK4: LearnReadOrientationModel | Gatk4LearnReadOrientationModel/4.1.8.1 |
| GATK4: MergeMutectStats | Gatk4MergeMutectStats/4.1.8.1 |
| GATK4: GetPileupSummaries | Gatk4GetPileupSummaries_cram/4.1.8.1 |
| GATK4: CalculateContamination | Gatk4CalculateContamination/4.1.8.1 |
| GATK4: GetFilterMutectCalls | Gatk4FilterMutectCalls/4.1.8.1 |
| BCFTools: Normalize | bcftoolsNorm/v1.9 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| normalBams | Array<CramPair> | The bams that make up the normal sample. Generally Mutect will expect one bam per sample, but as long as the sample ids in the bam header are set appropriatly, multiple bams per sample will work |
| tumorBams | Array<CramPair> | The bams that contain the tumour samples. Generally Mutect will expect one bam per sample, but as long as the sample ids in the bam header are set appropriatly, multiple bams per sample will work |
| normalName | String | The sample id of the normal sample. This id will be used to distingiush reads from this sample from all other samples. This id needs to tbe the one set in the bam header |
| biallelicSites | Gzipped<VCF> | A vcf of common biallalic sites from a population. This will be used to estimate sample contamination. |
| reference | FastaWithIndexes | A fasta and dict indexed reference, which needs to be the reference, the bams were aligned to. |
| panelOfNormals | Gzipped<VCF> | The panel of normals, which summarises the technical and biological sites of errors. Its usually a good idea to generate this for your own cohort, but GATK suggests around 30 normals, so their panel is usually a good idea. |
| germlineResource | Gzipped<VCF> | Vcf of germline variants. GATK provides this as well, but it can easily substituted with the newst gnomad etc vcf. |
| regionSize | Optional<Integer> | The size of the regions over which to parallelise the analysis. This should be adjusted, if there are lots of samples or a very high sequencing depth. default: 10M bp |
| createCallRegions_equalize | Optional<Boolean> |
Workflow Description Language¶
version development
import "tools/CreateCallRegions_v0_1_0.wdl" as C
import "tools/Gatk4Mutect2_cram_4_1_8_1.wdl" as G
import "tools/bcftoolsConcat_v1_9.wdl" as B
import "tools/bcftoolsIndex_v1_9.wdl" as B2
import "tools/Gatk4LearnReadOrientationModel_4_1_8_1.wdl" as G2
import "tools/Gatk4MergeMutectStats_4_1_8_1.wdl" as G3
import "tools/Gatk4GetPileupSummaries_cram_4_1_8_1.wdl" as G4
import "tools/Gatk4CalculateContamination_4_1_8_1.wdl" as G5
import "tools/Gatk4FilterMutectCalls_4_1_8_1.wdl" as G6
import "tools/bcftoolsNorm_v1_9.wdl" as B3
workflow Mutect2JointSomaticWorkflowCram {
input {
Array[File] normalBams
Array[File] normalBams_crai
Array[File] tumorBams
Array[File] tumorBams_crai
String normalName
File biallelicSites
File biallelicSites_tbi
File reference
File reference_fai
File reference_amb
File reference_ann
File reference_bwt
File reference_pac
File reference_sa
File reference_dict
Int? regionSize = 10000000
File panelOfNormals
File panelOfNormals_tbi
File germlineResource
File germlineResource_tbi
Boolean? createCallRegions_equalize = true
}
call C.CreateCallRegions as createCallRegions {
input:
reference=reference,
reference_fai=reference_fai,
regionSize=select_first([regionSize, 10000000]),
equalize=select_first([createCallRegions_equalize, true])
}
scatter (c in createCallRegions.regions) {
call G.Gatk4Mutect2_cram as mutect2 {
input:
tumorBams=tumorBams,
tumorBams_crai=tumorBams_crai,
normalBams=normalBams,
normalBams_crai=normalBams_crai,
normalSample=normalName,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
germlineResource=germlineResource,
germlineResource_tbi=germlineResource_tbi,
intervals=c,
panelOfNormals=panelOfNormals,
panelOfNormals_tbi=panelOfNormals_tbi
}
}
call B.bcftoolsConcat as concat {
input:
vcf=mutect2.out
}
call B2.bcftoolsIndex as indexUnfiltered {
input:
vcf=concat.out
}
call G2.Gatk4LearnReadOrientationModel as learn {
input:
f1r2CountsFiles=mutect2.f1f2r_out
}
call G3.Gatk4MergeMutectStats as mergeMutect2 {
input:
statsFiles=mutect2.stats
}
call G4.Gatk4GetPileupSummaries_cram as pileup {
input:
bam=tumorBams,
bam_crai=tumorBams_crai,
sites=biallelicSites,
sites_tbi=biallelicSites_tbi,
intervals=biallelicSites,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict
}
call G5.Gatk4CalculateContamination as contamination {
input:
pileupTable=pileup.out
}
call G6.Gatk4FilterMutectCalls as filtering {
input:
contaminationTable=contamination.contOut,
segmentationFile=contamination.segOut,
statsFile=mergeMutect2.out,
readOrientationModel=learn.out,
vcf=indexUnfiltered.out,
vcf_tbi=indexUnfiltered.out_tbi,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict
}
call B3.bcftoolsNorm as normalise {
input:
vcf=filtering.out,
reference=reference,
reference_fai=reference_fai
}
call B2.bcftoolsIndex as indexFiltered {
input:
vcf=normalise.out
}
output {
File out = indexFiltered.out
File out_tbi = indexFiltered.out_tbi
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: Mutect2 joint somatic variant calling workflow (CRAM)
doc: |-
This workflow uses the capability of mutect2 to call several samples at the same time and improve recall and accuracy through a joint model.
Most of these tools are still in a beta state and not intended for main production (as of 4.1.4.0)
There are also som major tweaks we have to do for runtime, as the amount of data might overwhelm the tools otherwise.
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
- class: ScatterFeatureRequirement
inputs:
- id: normalBams
doc: |-
The bams that make up the normal sample. Generally Mutect will expect one bam per sample, but as long as the sample ids in the bam header are set appropriatly, multiple bams per sample will work
type:
type: array
items: File
secondaryFiles:
- pattern: .crai
- id: tumorBams
doc: |-
The bams that contain the tumour samples. Generally Mutect will expect one bam per sample, but as long as the sample ids in the bam header are set appropriatly, multiple bams per sample will work
type:
type: array
items: File
secondaryFiles:
- pattern: .crai
- id: normalName
doc: |-
The sample id of the normal sample. This id will be used to distingiush reads from this sample from all other samples. This id needs to tbe the one set in the bam header
type: string
- id: biallelicSites
doc: |-
A vcf of common biallalic sites from a population. This will be used to estimate sample contamination.
type: File
secondaryFiles:
- pattern: .tbi
- id: reference
doc: |-
A fasta and dict indexed reference, which needs to be the reference, the bams were aligned to.
type: File
secondaryFiles:
- pattern: .fai
- pattern: .amb
- pattern: .ann
- pattern: .bwt
- pattern: .pac
- pattern: .sa
- pattern: ^.dict
- id: regionSize
doc: |-
The size of the regions over which to parallelise the analysis. This should be adjusted, if there are lots of samples or a very high sequencing depth. default: 10M bp
type: int
default: 10000000
- id: panelOfNormals
doc: |-
The panel of normals, which summarises the technical and biological sites of errors. Its usually a good idea to generate this for your own cohort, but GATK suggests around 30 normals, so their panel is usually a good idea.
type: File
secondaryFiles:
- pattern: .tbi
- id: germlineResource
doc: |-
Vcf of germline variants. GATK provides this as well, but it can easily substituted with the newst gnomad etc vcf.
type: File
secondaryFiles:
- pattern: .tbi
- id: createCallRegions_equalize
type: boolean
default: true
outputs:
- id: out
type: File
secondaryFiles:
- pattern: .tbi
outputSource: indexFiltered/out
steps:
- id: createCallRegions
label: Create genomic call regions
in:
- id: reference
source: reference
- id: regionSize
source: regionSize
- id: equalize
source: createCallRegions_equalize
run: tools/CreateCallRegions_v0_1_0.cwl
out:
- id: regions
- id: mutect2
label: GatkMutect2
in:
- id: tumorBams
source: tumorBams
- id: normalBams
source: normalBams
- id: normalSample
source: normalName
- id: reference
source: reference
- id: germlineResource
source: germlineResource
- id: intervals
source: createCallRegions/regions
- id: panelOfNormals
source: panelOfNormals
scatter:
- intervals
run: tools/Gatk4Mutect2_cram_4_1_8_1.cwl
out:
- id: out
- id: stats
- id: f1f2r_out
- id: bam
- id: concat
label: 'BCFTools: Concat'
in:
- id: vcf
source: mutect2/out
run: tools/bcftoolsConcat_v1_9.cwl
out:
- id: out
- id: indexUnfiltered
label: 'BCFTools: Index'
in:
- id: vcf
source: concat/out
run: tools/bcftoolsIndex_v1_9.cwl
out:
- id: out
- id: learn
label: 'GATK4: LearnReadOrientationModel'
in:
- id: f1r2CountsFiles
source: mutect2/f1f2r_out
run: tools/Gatk4LearnReadOrientationModel_4_1_8_1.cwl
out:
- id: out
- id: mergeMutect2
label: 'GATK4: MergeMutectStats'
in:
- id: statsFiles
source: mutect2/stats
run: tools/Gatk4MergeMutectStats_4_1_8_1.cwl
out:
- id: out
- id: pileup
label: 'GATK4: GetPileupSummaries'
in:
- id: bam
source: tumorBams
- id: sites
source: biallelicSites
- id: intervals
source: biallelicSites
- id: reference
source: reference
run: tools/Gatk4GetPileupSummaries_cram_4_1_8_1.cwl
out:
- id: out
- id: contamination
label: 'GATK4: CalculateContamination'
in:
- id: pileupTable
source: pileup/out
run: tools/Gatk4CalculateContamination_4_1_8_1.cwl
out:
- id: contOut
- id: segOut
- id: filtering
label: 'GATK4: GetFilterMutectCalls'
in:
- id: contaminationTable
source: contamination/contOut
- id: segmentationFile
source: contamination/segOut
- id: statsFile
source: mergeMutect2/out
- id: readOrientationModel
source: learn/out
- id: vcf
source: indexUnfiltered/out
- id: reference
source: reference
run: tools/Gatk4FilterMutectCalls_4_1_8_1.cwl
out:
- id: out
- id: normalise
label: 'BCFTools: Normalize'
in:
- id: vcf
source: filtering/out
- id: reference
source: reference
run: tools/bcftoolsNorm_v1_9.cwl
out:
- id: out
- id: indexFiltered
label: 'BCFTools: Index'
in:
- id: vcf
source: normalise/out
run: tools/bcftoolsIndex_v1_9.cwl
out:
- id: out
id: Mutect2JointSomaticWorkflowCram